
@Donald,
Thank you for the welcome note ![]()
Subsequent to the initial posts, have redesigned the entire thread.
IQVIA reports attached have been removed. The links are given in posts for people to download.
IQVIA has granted permission to share excerpts of the report by giving citation to the relevant reports - have followed that protocol.
Looking forward to discussions and interaction on the topic.
Thank you
Regards
Response to @pk89
-
Why are Chinese companies not aggressive in formulations? Very valid question! And I do NOT have any convincing answer on this one. From what I heard from my colleagues, they did not focus on International Generics opportunity - they found it easier to manage API.

The question was raised in a Pre-COVID-19 world. Post COVID-19, the Chinese folks won’t be seen through benign lens. I sense the era of distributed manufacturing (as compared to the existing concentrated manufacturing) is going to see sunrise now. -
Is the price erosion same for all types of products? Eg. Orals or injectables
A generic drug is a generic drug. Irrespective of the dosage form and route of administration.
Price Erosion is simply dependent on the number of competitors. The buyers are highly consolidated. The big suppliers are getting consolidated. So price erosion will always reach at the level where only the most cost-efficient suppliers for a drug remain in the market.
Please refer the example I gave in the earlier post:
Response to Donald
@Donald - My response is given in the following section.
Would like to know more from you about the Sun and Lupin success story in this area. Will be helpful if you can share some links to some articles on that? Or is it from there Annual Report or Conference Call of a particular period?
Drug Shortages in US Market
Federal Food, Drug, and Cosmetic Act (FD&C Act) - 21CFR - 356c.(h)(2) says:
The term “drug shortage” or “shortage”, with respect to a drug, means a period of time when the demand or projected demand for the drug within the United States exceeds the supply of the drug.
Lets take hypothetical example of Valsartan.
Assume that the annual demand (market volume) of Valsartan tablets (across all its strengths) is 500 Million Tablets in US.
Drugs@FDA database shows that following companies (15) have marketing approval for Valsartan:
• Novartis Pharmaceuticals Corp, • Alembic Pharmaceuticals Ltd, • Alkem Laboratories Ltd, • Amneal Pharmaceuticals of New York LLC, • Aurobindo Pharma Ltd, • Hetero Labs Ltd Unit V, • Ivax Pharmaceuticals Inc,• Jubilant Generics Ltd, • Lupin Ltd, • Macleods Pharmaceuticals Ltd, • Mylan Pharmaceuticals Inc, • Ohm Laboratories Inc, • Prinston Pharmaceutical Inc, • Square pharmaceuticals ltd, • Unichem laboratories ltd
Now due to the NDMA impurity issue in Valsartan’s API, Prinston (Solco / Zhejiang Huahai group) and several others had to recall the drug from the market and could not supply till the NDMA impurity is sorted out at API level.
This led to a situation where only those companies which used API sources that had synthesized API differently and did NOT have the NDMA impurity issue at API level could supply the formulation in US.
Lets assume that Alembic, Lupin and Jubilant were the only three entities which could sell drugs in the US market whereas all others were restricted till they resolve the NDMA impurity issue in the API.
Further - based on their existing API and Formulation capacities - lets assume that Alembic, Lupin and Jubilant can supply 200 Million, 150 Million and 100 Million Tablets of Valsartan in the next 1 year time horizon. [The corresponding API quantity - assuming 160 mg strength as weighted avg for entire Valsartan family - comes to 32 MT, 24 MT and 16 MT.]
This leads to a shortage of 50 Million Tablets annually [500 - (200 + 150 + 100)].
This being a situation where the existing API and formulation product is designated unsafe for human consumption, the channel inventory in US as well as inventories of formulation / API at manufacturers’ end (all those manufacturers who are undergoing the NDMA impurity issue) if of NO use. So the drug shortage is imminent.
What would happen in this scenario?
-
Formulation Manufacturers who are unable to supply due to API issue -
Will reach out to their API supplier (their own company dept. in case of vertically integrated folks) to get the NDMA impurity removed (or within the tolerable / permissible limits as the case maybe). -
API manufacturers facing the NDMA impurity will go back to basic chemistry of the drug and search for a synthesis method / route that successfully eliminates the problem. This will atleast take a few months - maybe more. The new route / process needs to be cost efficient as well. Perhaps not all API manufacturers can turn around with a new compliant process that gives Valsartan with the acceptable characteristics. Those who are able to get the desired output will have to file a new Drug Master File again. This entire process will definitely take minimum 1 year (as minimum of 6 months stability data needs to be included in the DMF submission).
-
Formulation manufacturers will use the new API and manufacture submission batches. But before they can ship the goods to US, they will most likely need to file Prior Approval Supplement (PAS) or if US FDA is benevolent enough then Changes Being Effected 30 (CBE-30) form with the US FDA.
PAS needs to be approved by US FDA (typical approval time is 4 months to 10 months - depending on the type of change filed) before the drug can be shipped to US. While CBE-30 is only a 30-day advance intimation/notification before shipping the drug product to US.
So depending on the case, at formulation end the restoration of supplies will take anything from 3 months to 1 year; making the minimum time of supply restoration - 15 months to 24 months (API + formulation). -
Coming over to folks who got their API chemistry right in the first attempt - Alembic, Lupin, Jubilant.
They get increased market share (now its a 3 player market for atleast 1.5 years) - between 40 percent to 25 percent for the companies - based on their throughput ability. Also they get far greater realization on their formulation supplies.
The increase in price could be well upto 10x. If the Net Selling Price for a bottle of 30s of 160 mg Valsartan was 1.5 Dollar earlier (basically around 5 cents per tablet) then the revised NSP could well be 15 Dollars per bottle (50 cents per tablet).
At a market volume of 450 Million - this translates into 225 Million Dollars per year molecule - to be shared between the three!!!
Perhaps they may not be able to capture the entire market volume in first year - if the batch sizes of API and formulation do NOT allow them to reach the required throughput. They may try to increase the batch size - at API as well as formulation level (if the constraint is present on both ends then on both manufacturing processes). Again, increase in batch size beyond a factor may require PAS so will not happen immediately.
Based on the above understanding regarding drug shortage caused due to API unavailability, lets explore drug shortage situations comprehensively. Lets build upon the content given in US FDA’s report on Drug Shortages : Root Causes and Potential Solutions - A Report by the Drug Shortages Task Force – 2019 – updated 21-Feb-2020.
Quoting from Pages 33 and 34 of the report:
When FDA adds a drug to the shortage list that it is required to maintain pursuant to the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA) it identifies one of the following reasons for the shortage:
- Requirements related to complying with good manufacturing practices
- Regulatory delay
- Shortages of an active ingredient
- Shortage of an inactive ingredient component
- Discontinuation of the manufacture of the drug
- Delay in shipping of the drug
- Demand increase for the drug, or
- Other reason
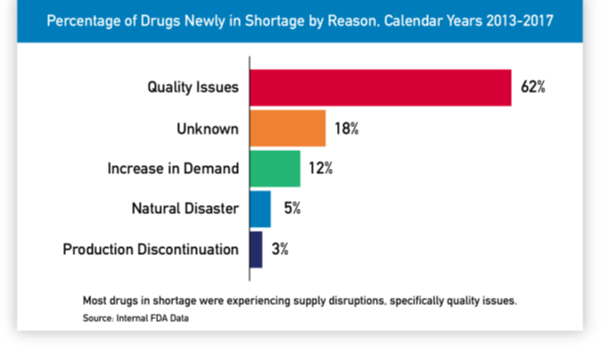
It is extremely essential to understand Point 2 (Regulatory delay) before we proceed further. Because timeline of most supply side issues is compounded and stretched due to the regulatory requirements involved in the process.
21 CFR 314.97 gives the reference of 21 CFR 314.70 for procedures relating to Supplements and other changes to an approved ANDA.
Quoting from Page 29 and Page 44 of the report:
-
In general, after a drug has been approved by FDA and is on the market, a manufacturer wishing to expand capacity through alternative suppliers (e.g., for API) or alternative manufacturing sites (e.g., for the finished product) must submit a regulatory filing to have the new supplier or manufacturing facility approved by FDA.
-
Many drug manufacturers supplying the U.S. market are in fact global operations that also supply other regions. Making post-approval changes to update manufacturing operations generally requires that they seek approval not only from FDA but the regulators in the other markets. According to industry observers, many post-approval changes to regulatory filings require prior approval by the regulatory authority of every country individually, and this can be over 100 countries for globally marketed products. The global approvals for changes can often take years because of varying requirements and timelines across different regulatory authorities, and this creates disincentives for timely improvements to manufacturing operations that could reduce the risk of drug shortages.
-
Under FDA regulations, a pharmaceutical firm making “major” manufacturing changes (which can include the remediation of a facility) must submit a Prior Approval Supplement (PAS). FDA approval of a PAS can take up to 4, 8, or 10 months depending on whether an inspection is required, and the applicant meets certain requirements.
-
By contrast, firms making “moderate” manufacturing changes must generally submit a Changes Being Effected in 30 days (CBE-30) supplement at least 30 days before the drug product is distributed. Changes that are moderate in nature and qualify for submission as a CBE-30 may be implemented by the sponsor after 30 days have passed and prior to completion of any review by the FDA. Because the CBE-30 process is much faster than gaining approval of the PAS, these industry representatives said it would be preferable to have more manufacturing changes made in response to a supply disruption handled through the CBE-30 pathway.
I would group the reasons for drug shortages as under:
- Supply side issues - Formulation level
-
Facility level import alert / ban - When the formulation facility fails to adhere to compliance / GMP and the company cannot supply products from that facility till FDA gives a green signal. This would generally impact supplies for > 6 months. Perhaps close to a year or more.
-
Change in formulation site / facility - If the product manufacturing is shifted to a manufacturing site having a different FEI (FDA Establishment Identifier) then PAS will get triggered. So minimum of 6 months time lag will occur before the supplies can begin from the different facility. During the transition phase if the organization is able to continue supplies from the existing facility then drug shortages and supply disruption may not occur. The facility change could be due to strategic de-risking, commercial considerations (lower conversion cost, proximity to the customer market) or technological superiority of the other site.
-
Batch failures - If few batches of a particular drug fail and the quantity is significant in relation to the market volume of drug then there can be temporary drug shortage in the market. Orphan Drug Products (serving to needs of <200,000 patients) are likely to see such scenarios. If the root cause of batch failure is such that remediation measure needs PAS then the drug shortage duration will extend to upto a year. If batch failures are remediated with CBE-30 or CBE-0 measures then the shortage remains only temporarily for 3-4 months.
-
Adverse drug reactions or effects - Formulation products for which significant / severe adverse drug reaction reports are received or there is safety concern raised by US FDA. The sartans NDMA incident will fall in this category. Root-cause identification and remediation will have to be done by the manufacturer. Again the remediation measures will need to be checked for PAS , CBE-30 or CBE-0. The timelines associated with resumption of supplies will depend on the regulatory pathway approved.
-
Transit damage / contamination - If a significant quantity of product was shipped together and there is transit damage or contamination which leads to product being rejected at US warehouse then there could be a temporary shortage in the market. Drug product shipments are done in temperature controlled environment and accompanied with Data-Loggers which keep a record of the environment condition (temperature, humidity etc.) in which the shipment is happening.
-
Changes in inactive substances, packing, labeling - Whether the changes is major or minor will be decided and in case FDA has directed the change as a major change then again PAS route needs to be taken.
-
Changes in manufacturing process / equipments / batch sizes - If there are changes made in manufacturing process, different set of equipments are used (which need to be validated for the drug product) or batch size is drastically changed (generally increased by 10x or more) then again regulatory clearance [PAS / CBE-30 or CBE-0] is required before the supply can be done. In cases where PAS is invovled, expect minimum 6 months time lag in supplies.
-
API source change (or new route API) - If API source is changed i.e. the API is sourced from a different FEI (FDA Establishment Identifier) than the current FEI. Also, cases where FEI is same but API has a new route DMF (adopted for commercial reasons like cost reduction or technical reasons). API from new FEI will mostly go through PAS route - so will easily need around 1 year for the change to be effected. New route API from same FEI may be allowed through CBE-30 route so can be done in a few months.
- Supply side issues - API Level
All the events which lead to non-availability of API at the formulation manufacturing facility. Major changes at API level, change in API facility site will need submission of a new DMF with 6 months of stability data.
-
Facility level import alert / ban on the API facility
-
Change in API manufacturing site / facility
-
Batch failures of API
-
Changes in manufacturing process / equipments / batch sizes of API
-
Chemistry level changes required in the basic synthesis of drug substance
- Demand surge in the market - There are several drug products which are prescribed for same disease indication and have the same mechanism of action. If we take the example of Sartans family [used for managing hypertension and are Angiotensin II Receptor Blockers (ARBs) in their mechanism of action] then patients will be prescribed that member of the sartans family which is best suited based on patients medical history and give minimum possible side-effects to a particular patient. Lets say Valsartan, Olmesartan and Telmisartan are the most prescribed drugs in this category. Now if US FDA receives significant adverse effect reports on Valsartan or finds that long term use of Valsartan is having unanticipated significant side-effects in a sizeable population of patients then it may decide to ban or suspend marketing of Valsartan. In such a scenario, as doctors start prescribing the other two sartan drugs (Olmesartan and Telmisartan) in place of Valsartan, the market size of Olme and Telmi will significantly increase in a short period of time.
Hope the above section throws adequate light on why drug shortages occur (the source of drug shortages) and why it takes time for the demand void to be filled in completely.
Coming to the other question raised by you:
why some businesses prove nimble enough to execute well on shortages more or less consistently; Sun and Lupin had supposedly mastered the Art? They had like real-time tabs on pharma stock-points across the country? Et al
Information / Visibility on drug products stock across the country -
There are two primary official sources of information:
- Industry databases (IQVIA, Bloomberg, Symphony, Integrichain etc.) provide weekly data on label-wise market share (label stands for the front-end entity which sells the drug products in US market) of each drug product.
- Channel Inventory (stock at Wholesalers / Distributors and Pharmacies) relating to drug products supplied by the front-end entity and consumption pattern/trend information is available from the distributors through EDI 852 report. They charge service fees for the information provided by them. Getting weekly EDI 852 reports will be costlier than getting monthly EDI 852 reports. The front-end entity decides the frequency of getting this information.
While 1. provides market share data of all labels selling a particular drug product, 2. provides channel inventory and consumption only related to sales made by the front-end entity.
The third source of channel inventory and warehouse stock levels of competitors is through unofficial channels ![]() Networking, relationships, unwritten quid-pro-quos will allow this information to flow.
Networking, relationships, unwritten quid-pro-quos will allow this information to flow.
Once a manufacturer is hit by an event which will lead to inability in supplying, the market share of its label will go down. But, due to channel inventory and warehouse inventory, this loss of market share might take 2-3 months to reflect in the published data. Unless ofcourse the situation relates to drug recall - then market share data will nosedive within 15 days.
Given the above facts, I don’t think that specific manufacturer(s) have positioned themselves to benefit from drug shortages. It is mostly a matter of lady luck smiling on the manufacturer(s). Once people sense an opportunity through the unofficial channels, they need to check with the plant sites about capacities, batch sizes, and raw material availability at formulation as well as API sites.
At best, as a measure of readiness, they would have validated products with large batch sizes at the time of commercial development and filing. Example - even if they know that the are likely to use commercial batch size of 500,000 tablets (say based on market share assumption of 10-12 percent), they would have done validation batch upto 10x i.e. 5,000,000 tablets / capsules so that when the opportunity comes they can do a quick turnaround.
Also we need to take cognizance of the fact that fulfilling 100% market demand may not be the best commercial decision from the manufacturer’s point of view. After they have supplied the immediate quantity relating to Failure-to-Supply fulfillment of the label which is unable to sell, they will watch the market actions of competitors and may decide to sell lower quantity at higher prices.
There are too many variables involved and the profits earned during drug shortages have to be attributed to luck - having the right capacities and material availability at the right time.
This is my take of the scenario. Happy to discuss further on this.
Thank you ![]()