Found an excellent summary of the FDA inspection process.
Recommend to visit the source URL for complete read.
http://www.metricstream.com/insights/FDA-inspections.htm
I have collected the important snippets for reference -
WHO DOES THE FDA INSPECT AND HOW OFTEN?
The Food, Drug, and Cosmetic (FD&C) Act gives FDA the authority to conduct inspections at drug and medical device manufacturing facilities, among other regulated types of facilities. The FDA selects companies to be inspected primarily based on risk . Those companies manufacturing drugs and higher risk devices, top the list. Thus, manufacturers of class III devices, sterile drugs, prescription drugs, newly registered facilities, implantable, life-supporting, and life-sustaining devices, are the primary subjects for inspection.
Facilities having historical significant violations, are also FDA inspected.
For pharmaceuticals and the risk based inspection system, FDA focuses on three types of facilities: sterile drug product manufacturers, those that produce other prescription drugs; newly registered facilities that had not been inspected previously ,and focuses on 3 factor categories: Quality and Product Safety; Facility; Process.
As per the Federal Food, Drug, and Cosmetics (FD&C) Act, domestic drug establishments, and Class II and Class III device manufacturers are to be inspected every two years (surveillance). Foreign manufacturers were inspected on an average of every 9 years. However, due to FDA’s budget increase, an increase in manufacturing plant inspections (domestic and international) has resulted. This budget increase is equipping the FDA to add staff and open international offices in major countries . Consequently, foreign inspections, do not have an every 9 year average anymore.
Inspections are also conducted prior to a company acquiring Premarket Approval Application (PMA) and New Drug Application (NDA) approvals, as well as Biologics License Application (BLA) licensing.
FDA QUALITY SYSTEM INSPECTION TECHNIQUE
FDA uses the Quality System Inspection Technique (QSIT) for its inspections. QSIT is based on a top-down approach to inspecting a manufacturer’s QS. The technique provides different inspectional levels based on reason for inspection.
In the medical devices industry, QSIT is used by the FDA to assess the firm’s QS for compliance with the appropriate regulations and for inspection of domestic and foreign manufactures of medical devices intended for commercial distribution in the United States.
The inspection will assess the firm’s systems, methods, and procedures to ensure that the firm’s quality management system is effectively established and maintained. The QS inspection should include the assessment of post-market information on distributed devices.
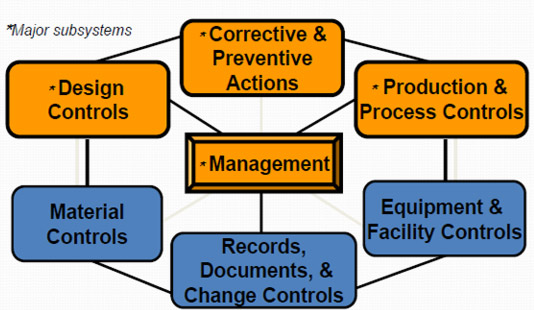
QSIT Devices Overview
The manner in which QSIT relates to the Pharmaceutical Industry, is through the guidance document: “Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations”. This guidance document defines requirements for all aspects of a firm’s quality structure. Firms that comply with the expectations of both 21 CFR Part 210/211 and QSIT, are aligned to the expectations of the referred guidance.
The Center for Drug Evaluation and Research (CDER) defines and groups QSIT into six major systems which consists of:

THE INSPECTION PROCESS IN A NUTSHELL
Here is what happens on the day of an FDA inspection:
• Receptionist activates the designated alert system.
• Company Inspection Team members go into their roles and designated areas.
• FDA provides the company with Form 482 and identification.
• At opening meeting, FDA explains why they are at your site.
• After a facility tour, the FDA inspection team, requests documents and begin to conduct interviews.
• FDA conducts the Exit Inspection Meeting.
• Findings are reviewed and description provided of any violations/deficiencies from current regulations, deviations from company’s own procedures and further clarification of misunderstandings can be addressed.
• FDA presents Notice of Observations (Form 483) to management and executive management present at exit meeting with annotations (the latter, if both parties agreed to annotations). The 483 lists inspectional observations. However, it does not represent a final Agency determination regarding your compliance.
• Upon return to local District Office, FDA investigator:
o Writes an Establishment Inspection Report (EIR) (normally, they begin writing it right at the firm’s site)
o Forwards report to headquarters and the Agency classifies the inspection
o After headquarter evaluates, you may receive a Warning Letter
WARNING LETTERS
If the inspection has an Official Action Indicated(OAI) classification, the FDA will send a warning letter to the company, describing the manufacturer’s violations of FDA regulations and requesting a reply, usually, within 15 working days after receipt of the letter. In such a case, the company will need to reply using the same format as the 483 response, and including documented evidence of corrective actions.
Usually , FDA conducts a follow-up inspection to verify that the appropriate corrections have been implemented and *are effective.
After the FDA has completed an evaluation of corrective actions via follow-up inspection, it may issue a clo*se-out letter.
A FEW BEST PRACTICES FOR SMOOTH FDA INSPECTIONS
Companies can ensure a smooth inspection process by following a few best practices:
• Have adequately trained resources with relevant expertise and accountability.
• Get upper management’s support on compliance and quality programs.
• Don’t underestimate the value of an independent regulatory compliance team and an expert quality assurance team.
• Make sure you have implemented the commitments made from previous inspection.
• Understand your potential quality data sources.
• Implement and assess an effective QS.
• Invest time, designate accountable resources.
• Ensure there is a designated company Inspection Team.
• Ensure proper documentation and records.
• Maintain effective Management Review and CAPA systems.
• Identify true root causes of issues using appropriate problem solving tools.
• In-bed in organizational culture, proactiveness.
• Understand when a product, or quality issue is significant.
• Have defined metric systems to monitor your QS in order to identify trends, gaps, and opportunities.
LEVERAGING TECHNOLOGY TO PREPARE FOR FDA INSPECTIONS
Both for 483s and warning letters, you will need to:
• Put together a project management team and assign accountability.
• Use change and document control systems, CAPAs and other quality systems to implement and monitor the changes needed and to sustain them.