
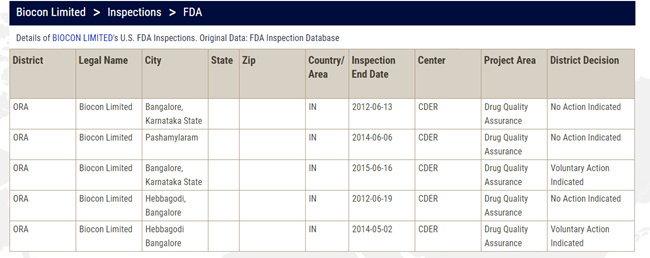
The inspection mentions date from March 27, 2017 to April 07, 2017. Surprisingly, we are knowing it now - a month later. Usually, the observations are handed over in a few days and companies are expected to make public.
The results came out around April 25. Does that mean they were aware of the observations then and did not make it public? If that is so, then why?
I am going through the list of observations. There doesn’t seem to be data integrity related issues. Will read and share details.
My concern is not what the observations are (they may be minor or major, doesn’t matter, as long as the management has the will to solve them), my concern is why were they not shared by the management with us and now why do we get to know of them from a third party (remember Divis Labs). My concern is that of transparency.
Management did admit during recent conference call that FDA inspection is over and as a policy they don’t comment on the outcome of inspections. They also said that the inspection result will not have any adverse effect on their US timeline although it sounded a little vague.
If they say it’s their policy not to comment on outcome of inspections, it is not a correct thing. Investors have a right to know what’s happening and the management is answerable. They cannot get away by saying it is a policy not to comment.
I have always held the Biocon management in high regard. And Biocon stands on a huge potential in next decade because of its niche products, both insulin, as well as oncology biosimilars. In fact, I purchased some, today morning, during the fall, at 995 (after seeing there is no specific data integrity issue in the observation; and seemingly not very serious, but to be safer side, brought only a very small quantity, till I understand the observations in detail). But I would have been happier if management had been transparent about all these issues. I find it discouraging if we come to know some news from a third party.
I have Read observations superficially. Per se they are not looking very serious. But I am not understanding a few points there. Will read in detail later and discuss here.
I agree with your view and also I tried making sense of the report-so not sure minor or major
My take on the FDA 483 - Clearly I was not expecting this, and although the observations are minor and easily rectifiable I would have assumed that Biocon would have had it’s act together knowing that an inspection was due. This was not a surprise inspection after all.
That being said, let’s put a couple of things in context -
-
About management policy of not commenting - They maintain their policy of not commenting on “non material” inspection outcomes. And based on the 483, the observations were indeed non material! How do I know this ? If the observations were material, there would be a temporary import freeze into the US on all products from said facility, until cleared after a re-inspection. This is clearly not the case, and business is going on as usual. So I have no quarrels with the management not commenting on this issue. Additionally they have unequivocally stated in the con call that this does not in anyway affect the timeline or anything else associated with their impending biosimilar decision dates later this year. So I’m not too worried.
-
Biocon has the best regulatory (FDA) track record, better than even several US companies. In the past 10 years (ending 2015) Biocon has received a grand total of 6 - 483 forms. This is miles ahead of most Indian and American companies including Mylan.
@Shivram - Differentiating on quality based on the end destination of the drug is routine practice. That;'s how the industry operates. The compliance cost for drugs manufactured for the US & EU markets are extremely high, No pharma company would maintain US standards and incur the compliance costs for drugs to be marketed in Africa for instance. The selling price of the drug in Africa, as an example, would not cover the additional compliance costs incurred. The determining factor here is country specific compliance requirements. So not sure what you mean by saying “they should not differentiate and quality should be a culture” That’s how the industry works, and for good reason.
In conclusion, I’m disappointed that the FDA did find Biocon lacking and made observations requiring additional management action, but that being said, it changes nothing in so far as my investment thesis goes. September & October of this year are extremely important for Biocon & that’s all that I’m focused on.
Note- I am extremely biased, and happy to admit it. So please take everything I say with a spoonful of salt. But my focus is only on the biosimilar business & so anything that does not in anyway affect/delay/derail that is just noise to me.
To state that US FDA observations are not material is, in my opinion,a serious governance lapse. Does that mean the company is not taking these observations seriously as it is immaterial? If the company is serious about these observations why is it focussing on it being immaterial - how does that matter at all - even one observation means there is something wrong that needs to be corrected immediately, not responded to. It is a fact that the company will have and has no idea how US FDA is going to react (follow Divi’s lab for instance). Rather than arbitrarily decide that there is no material impact, Biocon management would have done well to disclose that there were US FDA observations for which they have responded and that they believe these will not cause significant impact to the business. I am not entirely convinced that there is no chance for an import alert here.
I sold my holdings in Biocon. I agree with Rohit that the most important events are in Sep and Oct. But I would rather wait to make sure that there are no risks to these events rather than hope that all is well.
Also as regards my point on quality, while it is true that the industry operates by providing better quality to developed markets and sub-par quality to the rest, this does not seem to augur well in the long term. Invariably the companies seem to perform as well as its weakest link and therefore get into trouble with FDA authorities. According to an interpretation of Porter (by Joan Magretta), lasting competitive advantage is always with a company which is able to configure its activities in such a way that it is able to deliver products / services to its segment market at a low cost or a premium price in a manner which is difficult to imitate. And companies which try to configure this in multiple ways to target multiple customer segments at the same time lose this edge because this is simply impossible to do (British airways failed miserably when it tried to manage a low cost airline too). I am afraid Indian Pharma companies are losing their way here trying to be everything to everyone.
US FDA observations are not significant. Q4 numbers were underwhelming. Hence the sentiment is negative and stock price has corrected. Once the trend reverses I will invest more money. Long term growth is intact in my view.
firstly, let me record my appreciation for the quality of discussion that this forum has generated, especially the initial introduction from rks00. I have just now joined the forum after observing it from the outside for a good amount of time.
I will make these initial points before elaborating on them as we take this discussion forward:
- the Biocon story has a ‘long’ way further to play out, and
- some of the management commentary on ROE and Capex budgets (for the next 3-5 years) point towards significant revenue & profit growth from FY 18-19 onwards.
Roche Aphinity trial, results out at ASCO 2017. Addition of Perjeta shows only marginal improvement in survival as compared to herceptin alone (94 vs 93 %). Bodes well for Trastuzumab biosimilars.
I have a slight difference of opinion : According to NEJM publication of Aphinity:
The study was designed for a HR of 0.75 using iDFS (standard primary entry point). The control arm was expected to reach 89.2% & it actually did better at 93.2%…In fact MS (like lower cardio toxicity etc.) report has few more points from the primary investigator indicating that it contributes to de-risking of Roche’s breast cancer franchise
http://www.nejm.org/doi/full/10.1056/NEJMoa1703643#t=article
Also Mylan’s FDA panel on trastuzumab is on 13th July afternoon session
FDA link: http://bit.ly/2qSHuvX
While dissecting the Aphinity trial and arriving at a definitive conclusion is beyond me, here is an accompanying editorial in NEJM that paints a not so favourable opinion on the said trial.
If you would read it , the article that I have mentioned is presented by 10+ experts in response to the article that you have posted (written by Kathy MIller). Again not being an expert , I am inclined to believe that article that I have posted has larger universe of researchers who have presented their response to Kathy MIller. The point I was trying to make is that Roche can spring a surprise and its not an open field for Trastuzumab. I have no exposure to Biocon & dont intend to take.
As far as treatment is concerned, breast cancer can be grouped in to three.
Neoadjuvant: where you give chemotherapy(CT) prior to surgery to downgrade the tumour so that adequate resection is possible
Adjuvant: Where CT is given after surgery, with the goal of decreasing tumour reccurrence.
Metastatic: Where CT is given to take care of tumour that has already spread to distant organs
Of these 3, Adjuvant group is the largest( 60% ) while Neoadjuvant (10%) and metastatic(20%) account for the rest.
Perjeta in combination with trastuzumab and docitaxel is the current standard of care in both neoadjuvant and metastatic settings based on studies that showed a marked advantage when pertuzumab was added to standard CT. So 30% of breast cancers is already excluded from serious biosimilar competition.
With Aphinity trial, it was hoped that perjeta combination would prove to be markedly superior to trastuzumab in adjuvant setting also; thereby bringing almost 90% of breast cancers under the Roches umbrella .
This didnt materialize.
Addition of perjeta caused a less than 1 % survival advantage in adjuvant setting, that too at the expense of double the cost and significant side effects like diarrhea and cardiotoxicity.
So although Aphinity is definitely a positive trial, the quantum of advantage is not significant enough to make perjeta standard of care in adjuvant setting (my assumption). This leaves the largest pie of breast cancer still open to competition by biosimilars.
Looks like I have crossed the line  Dr. Could not understand most of your stuff -dont have your level of knowledge on treatments -thank you anyway for taking discussion to a higher level. Just my penny’s worth
Dr. Could not understand most of your stuff -dont have your level of knowledge on treatments -thank you anyway for taking discussion to a higher level. Just my penny’s worth
The cardio toxicity apparently came in as expected & Diarrhea was dependent on the type of chemotherapy
Also read a broker’s note on some physicians at ASCO (again a mixed review) indicating that they would use Perjeta in node positive patients & HR (hormone receptor) negative patients. Point taken on what you written. Biocon has a wide field to play without Roche playing spoil sport. I have no exposure & dont intend to take
Have been with BIOCON for years now and always out-of-breath. As complex a company’s
as it could get. Appreciate your and doctors inputs.
Mylan (MYL) & Biocon Ltd. Presented New Clinical Data on Insulin Glargine Program at ADA Meetings
https://www.streetinsider.com/dr/news.php?id=13000343
Arun Chandavarkar, CEO & Joint Managing Director, Biocon, added, “We are pleased with the outcome of these global clinical studies confirming the safety, efficacy and immunogenicity of our insulin glargine in comparison to the reference product in Type 1 and Type 2 diabetes. This is an important milestone in our development of a more affordable insulin glargine and furthers our mission of enabling access by addressing the needs of diabetes patients globally.”
Thanks
Mylan-Biocon’s biosimilar of Amgen’s Neulasta, a longer-acting form of Neupogen that prevents infection in cancer patients, faces a high hurdle for FDA approval. The copy is slated to be reviewed by the agency by Oct. 9, though past reviews have indicated that the demonstration of biosimilarity is difficult for this molecule, likely due to the addition of a molecule called PEG that slows the metabolism of the drug to make it longer acting. If approved, it would be the first, given peers have been delayed-BI specialty -generic pharma dashboard. They seem the first off the block considering Coherus application is delayed & Sandoz’s application was rejected.
The latest verdict by US Supreme Court in the case of Amgen versus Sandoz will help biosimilar makers expedite the launch of the lifesaving drugs without waiting an additional six months after exclusivity period ends.
Biologics Price Competition and Innovation Act (BPCIA) requires biosimilar drug developers to give reference drug makers 180 days’ notice that they will launch a biosimilar version. The 180 days provision gives six extra months of exclusivity over and above the 12-year exclusivity granted to reference drug developers under the BPCIA,
positive news for biosimilar market may be BPCIA’s “patent dance” can be avoided
thanks
Found this while trying to understand the lifecycle of biosimilars vis-à-vis the patent dance.